Structure D’une Protéine R2 De La Ribonucléotide Réductase Avec Un Radical.
Production et purification des protéines
La souche Mesoplasma florum R2 a été produite et purifiée en utilisant le plasmide pET28MfnrdFI, comme décrit précédemment. En bref, les gènes nrdF et nrdI de M. florum ont été amplifiés par PCR à partir de l’ADN génomique de M. florum L1 (NCTC 11704, obtenu auprès de Public Health England). Tous les codons traduisant en tryptophane dans le gène nrdF ont été mutés de TGA à TGG pour corriger la différence de l’utilisation des codons entre M. florum et Escherichia coli. Les gènes nrdF et nrdI ont été clonés dans le plasmide pET-28a (Novagen) précédemment modifié, avec le site de clivage par la thrombine remplacé par un site de clivage par la protéase du virus de la mosaïque du tabac (TEV). Le plasmide généré, pET28MfnrdFI, comporte le gène nrdF directement en aval de la 6xHis-tag clivable par la TEV, suivi d’un site de liaison au ribosome devant le gène nrdI. E. coli BL21(DE3) (NEB) portant le plasmide pRAREcamR (Novagen) a été transformé avec le plasmide pET28MfnrdFI. Les cultures ont été cultivées dans un milieu LB (Formedium) à 37 °C, supplémenté de kanamycine et de chloramphénicol, dans un système de bioréacteur de paillasse. Lorsqu’une densité optique à 600 nm de 0,7 a été atteinte, l’expression a été induite avec 0,5 mM d’isopropyl β-D-1-thiogalactopyranoside et autorisée à se poursuivre toute la nuit à température ambiante. Ensuite, les cellules ont été récoltées par centrifugation et stockées à -80 °C. La protéine MfR2 marquée par la 6xHis a été purifiée à 4 °C par chromatographie d’affinité par immobilisation des ions métalliques (IMAC) et par chromatographie par exclusion de taille (SEC). Un culot cellulaire a été resuspendu dans un tampon A (25 mM de HEPES-Na pH 7.0, 20 mM d’imidazole et 300 mM de NaCl) et a été rompu par homogénéisation à haute pression (EmulsiFlex-C3). Le lysat a été clarifié par centrifugation, appliqué sur une colonne à gravité d’agarose à nitrilotriacétate de nickel et lavé abondamment avec un tampon B (tampon A contenant 40 mM d’imidazole). La protéine a été éluée en utilisant un tampon C (tampon A contenant 250 mM d’imidazole), concentrée en utilisant des concentrateurs centrifuges Vivaspin (poids moléculaire de coupure de 30 kDa, Sartorius), et appliquée sur une colonne SEC HiLoad 16/60 Superdex 200 prep grade (GE Healthcare) équilibrée dans un tampon final de 25 mM de HEPES-Na pH 7.0 et 50 mM de NaCl. Les fractions correspondant à MfR2 ont été regroupées et soumises à une clivage par la protéase TEV. Une étape d’IMAC inverse a été réalisée pour isoler la MfR2 sans étiquette 6xHis, qui a ensuite été concentrée. La pureté de la protéine et la teneur en radical ont été vérifiées par SDS-PAGE et spectrophotométrie UV-visible. Enfin, la protéine a été aliquotée, congelée rapidement et stockée à -80 °C.
Quenching du radical MfR2 par le traitement à l’hydroxyurée
Avant la cristallisation, 50 μl de MfR2 actif, porteur de radicaux bleus, à une concentration de 0,6 mM, ont été incubés pendant 24 heures à température ambiante avec 100 mM d’hydroxyurée ou d’urée. L’hydroxyurée est un éliminateur de radicaux RNR connu. L’urée a été utilisée comme témoin, pour confirmer que l’étape de quenching des radicaux n’était pas le résultat d’une dénaturation non intentionnelle de la protéine par l’hydroxyurée. Ensuite, l’hydroxyurée et l’urée ont été éliminées par des étapes d’échange de tampon sur un concentrateur Vivaspin Turbo 4, coupé à un poids moléculaire de 30 kDa (Sartorius), et MfR2 a été concentré à 25 mg ml−1 pour la cristallisation. La protéine a conservé sa couleur bleue après l’incubation avec l’urée, mais est devenue incolore lorsqu’elle a été incubée avec l’hydroxyurée, confirmant que le quenching des radicaux s’est produit uniquement lors du traitement à l’hydroxyurée.
Cristallisation de MfR2 dans l’état sans radical pour la collecte de données de synchrotron
MfR2 a été cristallisé à une concentration de 25 mg ml−1 en utilisant la méthode de diffusion de vapeur par goutte suspendue dans des plaques de cristallisation MRC à 2 puits à l’aide d’un robot de pipetage nanolitre mosquito (TTP Labtech) et un volume de réservoir de 50 μl. La condition du réservoir était de 175 mM d’acétate de calcium, 100 mM de sulfate d’ammonium et 20% (p/v) de polyéthylène glycol (PEG) 3350. Le rapport entre la protéine et le volume du réservoir était de 0,2:0,2 μl. Des cristaux cuboïdaux de couleur bleue ont poussé en 2 jours à 21 °C. Pour accélérer le processus de cristallisation, des microcristaux ont été ajoutés à la goutte de cristallisation. Un stock de microcristaux a été préparé en resuspendant un cristal de MfR2 dans un tube de 1,5 ml contenant 50 μl de solution de réservoir et une bille PTFE de 3,2 mm (Saint-Gobain) maintenue sur glace, et en l’écrasant par des cycles de vortex successifs de 30 s. Pour MfR2 traité à l’hydroxyurée, des cristaux cuboïdaux incolores apparaissent en 24 heures à 21 °C (dimensions d’environ 100100100 μm3). Avant la collecte de données sur une source de synchrotron, les cristaux ont été cryoprotégés avec une solution composée de solution de réservoir supplémentée de 20% de glycérol et congelés rapidement dans de l’azote liquide.
Collecte de données, réduction des données et détermination de la structure par synchrotron
Les données de diffraction des rayons X de synchrotron ont été collectées à 100 K à la ligne de faisceau X06SA (PXI) de la Swiss Light Source (Paul Scherrer Institute, Villigen, Suisse). Des données de diffraction des rayons X complémentaires ont été collectées sur les lignes de faisceau PROXIMA 1 et PROXIMA 2A du synchrotron SOLEIL (Saint-Aubin, France). La réduction et la mise à l’échelle des données ont été effectuées à l’aide de XDS. La coupure en haute résolution de l’ensemble de données correspondant à l’état sans radical de MfR2 a été déterminée sur la base d’une combinaison de I/σ(I), Rmeas et CC1/2. Le cristal appartient au groupe d’espace C2 avec des cellules unitaires similaires aux structures MfR2 que nous avons précédemment résolues. Les phases ont été récupérées en effectuant une réaffinement de corps rigide dans phenix.refine en utilisant un modèle édité avec des facteurs B réinitialisés sur les coordonnées atomiques de la forme DOPA de MfR2 (PDB ID: 6gp2), définissant chacune des deux chaînes présentes dans l’unité asymétrique comme des groupes distincts.
Cristallisation pour la collecte de données XFEL
Afin de fournir un nouveau stock de microcristaux, MfR2 a été cristallisé en utilisant la méthode de diffusion de vapeur par goutte suspendue dans des plaques de cristallisation MRC Maxi (Swissci), en mélangeant un volume de 2 μl d’une solution de protéine à 25 mg ml−1 avec 2 μl de solution de réservoir composée de 200 mM d’acétate de calcium, 10 mM de sulfate d’ammonium et 20% (p/v) de PEG 3350. La solution de stock de microcristaux a été préparée comme décrit précédemment. Les microcristaux utilisés pour la collecte de données XFEL ont été cultivés en utilisant la méthode en lot dans un tube contenant 12 μl de solution MfR2 à 13 mg ml−1, 12 μl de tampon de cristallisation et 0,8 μl de solution de stock de microcristaux pour une formation rapide de cristaux. Le tampon de cristallisation était composé de 50 mM d’acétate de calcium, 20 mM de sulfate d’ammonium et 20% (p/v) de PEG 3350. Les tubes ont été incubés verticalement à 21°C pendant 24 heures, permettant la croissance de cristaux en forme de plaques (dimensions d’environ 10105 μm3), avant d’être regroupés, resuspendus dans un tampon de cristallisation supplémenté de 10% (v/v) de glycérol, et chargés dans une seringue Hamilton maintenue sous rotation à 21°C.
Livraison d’échantillons XFEL (Free Electron Laser) et collecte de données
Les données XFEL ont été collectées sur l’instrument de cristallographie macromoléculaire à femtoseconde (MFX) du Linac Coherent Light Source (LCLS) de la SLAC National Accelerator Laboratory, aux États-Unis, en utilisant des impulsions de rayons X d’une durée de 35 fs à une énergie de 9,5 keV, avec une fréquence de répétition de 30 Hz, une puissance moyenne d’impulsion de 2,5 mJ et une taille de faisceau de 4 μm (FWHM), obtenue par des lentilles réfractives composites. L’échantillon a été livré à température ambiante sous forme de petites gouttes de suspension cristalline générées par éjection acoustique de gouttelettes depuis un petit réservoir, puis transporté dans la région d’interaction des rayons X par une ceinture de polyimide à l’intérieur d’une enceinte d’hélium, en utilisant le dispositif « Drop on Tape » décrit précédemment. En résumé, la suspension cristalline s’écoulait en continu depuis une seringue Hamilton étanche de 1 ml dans un réservoir d’échantillon de 6 μl situé sous un transducteur acoustique focalisé. La suspension d’échantillon était livrée à un débit d’environ 9 μl/min. Des gouttelettes de 5 nl de volume ont été générées à l’aide d’un transducteur de 10 MHz (pour plus de détails et éjectées à 30 Hz en synchronisation avec la lecture du détecteur et la livraison de l’impulsion XFEL. La ceinture de polyimide fonctionnait à une vitesse de 300 mm/s, exposant les gouttelettes formées à l’atmosphère d’hélium de l’enceinte pendant 0,8 s avant d’être sondées par l’impulsion de rayons X. Les données de diffraction ont été détectées à une fréquence de 30 Hz sur un détecteur Rayonix MX340-HS en mode binning 4 x 4 à une distance de 144 mm du point d’interaction. Les données ont été collectées pendant 35,2 minutes, produisant 63350 clichés aux rayons X, dont 14431 ont été indexés (taux d’indexation de 23%).
Réduction des données XFEL et détermination de la structure Les données XFEL ont été traitées à l’aide de cctbx.xfel et DIALS comme décrit précédemment. Avant le traitement, nous avons appliqué une révision conjointe du modèle de détecteur Rayonix sur 799 modèles cristallins (dans ce cas, provenant d’un échantillon différent collecté simultanément avec R2e), générant une nouvelle position du détecteur. Même avec cette position affinée, pour ces données, nous avons initialement eu du mal à déterminer le groupe spatial et la maille unitaire. En bref, nous avons indexé les données en utilisant une maille unitaire cible initiale de a = 176,2 Å, b = 53,6 Å, c = 79,2 Å, α = γ = 90°, β = 108,6° et un groupe spatial C2, car il s’agissait de l’isoforme présumée des cristaux. De manière inattendue, la maille cristalline semblait être différente, comme suggéré par le faible taux d’indexation des images cristallines. La retraitement des données en P1 sans maille cible a amélioré le taux d’indexation cinq fois. Après avoir utilisé le programme cluster.unit_cell pour identifier les cellules probables en fonction du regroupement en P1, nous avons reconnu que la maille avait une similitude avec un ensemble de données synchrotron non publié en C2, avec une maille de a = 143 Å, b = 46 Å, c = 58 Å, α = γ = 90°, β = 113°. En utilisant cela comme cible d’indexation, le taux d’indexation s’est amélioré de 15%, et à partir de ces résultats, nous avons pu résoudre la structure. Nous avons fusionné 14431 images à une résolution de 1,50 Å en utilisant cxi.merge, faisant partie du package cctbx.xfel. Pour réduire les effets de la fusion du signal non mesuré, nous avons calculé une coupure de résolution pour chaque image en utilisant des estimations I/σ(I) pour déterminer les limites de résolution de chaque cristal (les erreurs ont été estimées en utilisant le modèle Ha14). Nous avons également appliqué une post-raffinement pour chaque image, dans lequel l’orientation du cristal, le facteur B de Wilson et le facteur d’échelle sont raffinés, la différence entre les intensités de réflexion corrigées partiellement échelonnées et les intensités du modèle. La correction de partialité gonfle les intensités de réflexion à leurs équivalents complets, analogues aux intensités qui auraient été mesurées si le cristal avait été tourné à travers la sphère d’Ewald. La structure cristalline a été résolue à l’aide de Phaser par substitution moléculaire en utilisant comme modèle de recherche la chaîne A des coordonnées atomiques de la forme DOPA de MfR2 (PDB ID: 6gp2). Une solution bien contrastée a été obtenue avec une molécule par unité asymétrique dans le groupe spatial C2.
Raffinement du modèle des structures synchrotron et XFEL
Tous les modèles cristallographiques ont été examinés et modifiés à l’aide du programme Coot et raffinés à l’aide de phenix.refine dans la suite PHENIX. Le raffinement du facteur B a été effectué en utilisant des paramètres de déplacement atomique (ADP) anisotropes individuels pour les atomes de protéine et de ligand, et des ADP isotropes pour les molécules d’eau. La validation de la structure a été effectuée avec MolProbity. Des cartes de densité électronique composites Omit 2Fobs–Fcalc simulées par recuit ont été générées pour les structures MfR2e dans les états radical et radical-lost en utilisant phenix.composite_omit_map. La méthode « anneal » a été choisie avec 5% du modèle omis à chaque étape et les réflexions Rfree supprimées. La Table a été générée avec phenix.table_one et liste les statistiques cristallographiques où l’ensemble de test représente 5% des réflexions. Les statistiques Ramachandran sont les suivantes : favorisées 99,01%, et 99,34% pour les structures cristallines MfR2 de l’état radical et de l’état fondamental radical-lost, respectivement. Les valeurs aberrantes de Ramachandran étaient de 0,0% dans les deux structures. Les représentations des surfaces intérieures ont été générées à l’aide de HOLLOW. La conservation des résidus a été obtenue grâce aux alignements de 8924 séquences R2 uniques provenant de la base de données NCBI RefSeq, décembre 2018 (une séquence par espèce et sous-classe). Nous notons que la protéine produite selon le protocole décrit a une teneur en radical d’environ 52%. Cependant, dans la structure de l’état radical, nous observons une seule conformation bien définie. Il semble donc que l’état conformationnel radical a été préférentiellement sélectionné lors de la cristallisation.
Analyse d’erreur END/RAPID
La méthode END/RAPID a été utilisée pour estimer la précision de l’attribution de la distance entre les atomes d’oxygène de DOPAY126 et D88. L’idée centrale derrière la méthode END/RAPID est que nous voulons décrire les conséquences de la répétition de la même mesure de données structurales plusieurs fois en termes du modèle structural résultant et en déduire la fiabilité de différents aspects du modèle final dérivé. Pour simuler la répétition de l’expérience de diffraction, nous introduisons des erreurs aléatoires aux mesures d’intensité (basées sur les estimations d’erreur pour les intensités) et effectuons une raffinement structural séparée pour chacun de ces ensembles d’intensités perturbées, générant un modèle structural raffiné différent. Lorsque cette procédure est répétée plusieurs fois, les différences entre les différents modèles structuraux capturent adéquatement l’erreur de modélisation causée directement par les erreurs dans les intensités mesurées. La méthode END/RAPID rend ainsi possible la propagation des erreurs de mesure d’intensité aux erreurs du modèle structural pour les macromolécules. Dans ce travail, nous avons d’abord perturbé les facteurs de structure échelonnés (Fobs échelonnés à Fcalc) d’une quantité aléatoire dans la plage de ± |Fobs – Fcalc| (en supposant que l’erreur maximale des mesures est similaire à l’écart entre les facteurs de structure observés et calculés) pour 100 essais distincts. Cela nous donne 100 ensembles de données indépendants pour le raffinement. Étant donné que la modélisation des erreurs d’intensité pour les expériences XFEL est encore un domaine de recherche actif, nous avons choisi d’utiliser la plage de ± |Fobs – Fcalc| car elle reflète une borne supérieure pour les erreurs de mesure possibles. Ensuite, pour chacun des 100 essais distincts, nous avons également perturbé le modèle structural de départ d’une quantité aléatoire en utilisant l’outil sites.shake dans Phenix. Avant de raffiner contre les données perturbées, les contraintes pour les molécules de DOPA ont également été assouplies (0,2 Å pour les distances de liaison et 20 degrés pour les angles). Ensuite, nous avons effectué des travaux de raffinement indépendants pour obtenir 100 modèles structuraux raffinés. Les paramètres de raffinement pour ces ensembles de données perturbés ont été maintenus les mêmes que pour le raffinement original, à l’exception des contraintes plus lâches de la molécule de DOPA, et le même logiciel de raffinement a été utilisé. À partir de l’ensemble de modèles raffinés, nous avons calculé l’écart type de la distance entre DOPAY126 méta-O (OE2) et OD2 de D88. Cet écart type a été rapporté dans le texte principal.
Nous notons que, parce que l’amplitude de la perturbation des facteurs de structure est plus grande que les vraies erreurs expérimentales, nous nous attendons à ce que l’estimation d’erreur obtenue pour les distances soit une borne supérieure de l’erreur réelle.
Pour compléter l’approche END/RAPID, les erreurs de longueur de liaison (σl) entre le méta-O (OE2) de DOPAY126 et OD2 de D88 ont été calculées en utilisant l’équation (4) de la référence (63) : σl = (σ2a + σ2b)1/2. Les erreurs de coordonnées atomiques (σa et σb) ont été calculées à l’aide du serveur d’index de précision de diffraction Online_DPI. Pour les structures de l’état radical et de l’état fondamental radical-lost, les erreurs de longueur de liaison sont de 0,09 Å et 0,06 Å, respectivement. Les deux valeurs sont comparables (bien que plus élevées) avec les valeurs des écarts types obtenus à l’aide d’END/RAPID (0,05 Å et 0,04 Å, respectivement).
Détails computationnels
Les calculs quantiques chimiques ont été réalisés en utilisant la DFT avec la fonction B3LYP-D3 et la suite Jaguar de Schrödinger (Schrödinger Release 2018-3: Jaguar, Schrödinger, LLC, New York, NY, 2018). Un modèle du site actif a été construit en incluant les résidus. Les géométries ont été optimisées sous vide avec l’ensemble de base 6-31g**, en maintenant les alpha-carbones et les cap-protons le long du squelette fixés. Les énergies ponctuelles ont été calculées sous vide avec l’ensemble de base cc-pvtz(-f), et les effets de solvatation issus d’un calcul pbf dans un milieu protéique avec une constante diélectrique de 4 et l’ensemble de base 6-31g** ont été ajoutés.
Des simulations de dynamique moléculaire (MD) ont été réalisées pour explorer la dynamique conformationnelle de l’état perdu du radical de DOPAY126. À cette fin, un modèle de départ obtenu à partir de données XFEL (état radical) du MfR2 dimérique a été intégré dans une boîte d’eau (TIP3P) avec 150 mM de NaCl, et simulé dans l’état perdu du radical en utilisant le champ de force CHARMM36. Le système, comprenant 83 435 atomes, a été simulé en triplicata dans un ensemble NPT, avec p = 1 atm et T = 310 K en utilisant un pas de temps d’intégration de 1 fs, et en traitant les interactions électrostatiques à longue portée avec l’approche Particle Mesh Ewald. Les simulations de MD ont été effectuées avec NAMD v. 2.14/3.0.
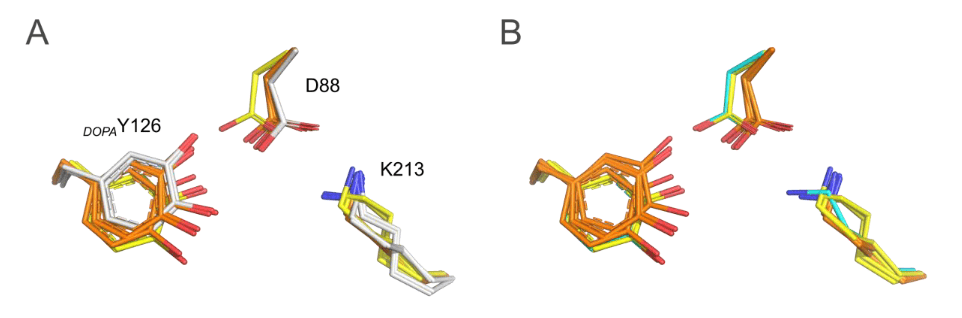
Image 1. Superposition des structures de MfR2 dans les états radical et radical-lost avec les structures de R2e précédemment publiées de manière synchrotron pour Aerococcus urinae (AuR2, ID PDB : 6ebq) (14) et Mesoplasma florum (MfR2, ID PDB : 6gp2). Cette comparaison met en évidence que toutes les structures de R2e précédemment publiées, cristallisées initialement dans la « forme active », diffèrent significativement des états radical et radical-lost résolus dans cette étude, très probablement en raison de la photoréduction induite par les rayons X pendant la collecte de données synchrotron et/ou de l’occupation initiale partielle de l’état radical. Pour plus de clarté, les figures incluent uniquement trois résidus clés (numérotation de MfR2) et aucune molécule d’eau, mais d’autres différences et conformations alternatives peuvent être observées dans l’ensemble des structures. (A) MfR2 dans l’état radical-lost (gris, chaînes A et B) comparé aux structures synchrotrons précédemment publiées d’AuR2 (orange, chaînes A à D) et de MfR2 (jaune, chaînes A et B). (B) MfR2 dans l’état radical (cyan, chaîne A) comparé aux structures synchrotrons précédemment publiées d’AuR2 (orange, chaînes A à D) et de MfR2 (jaune, chaînes A et B).
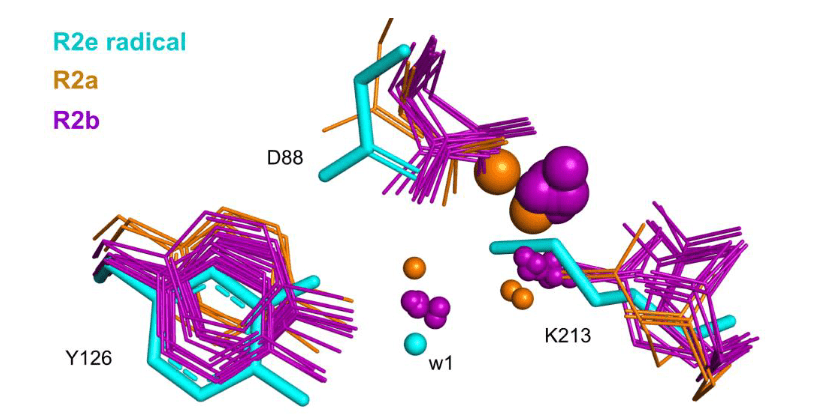
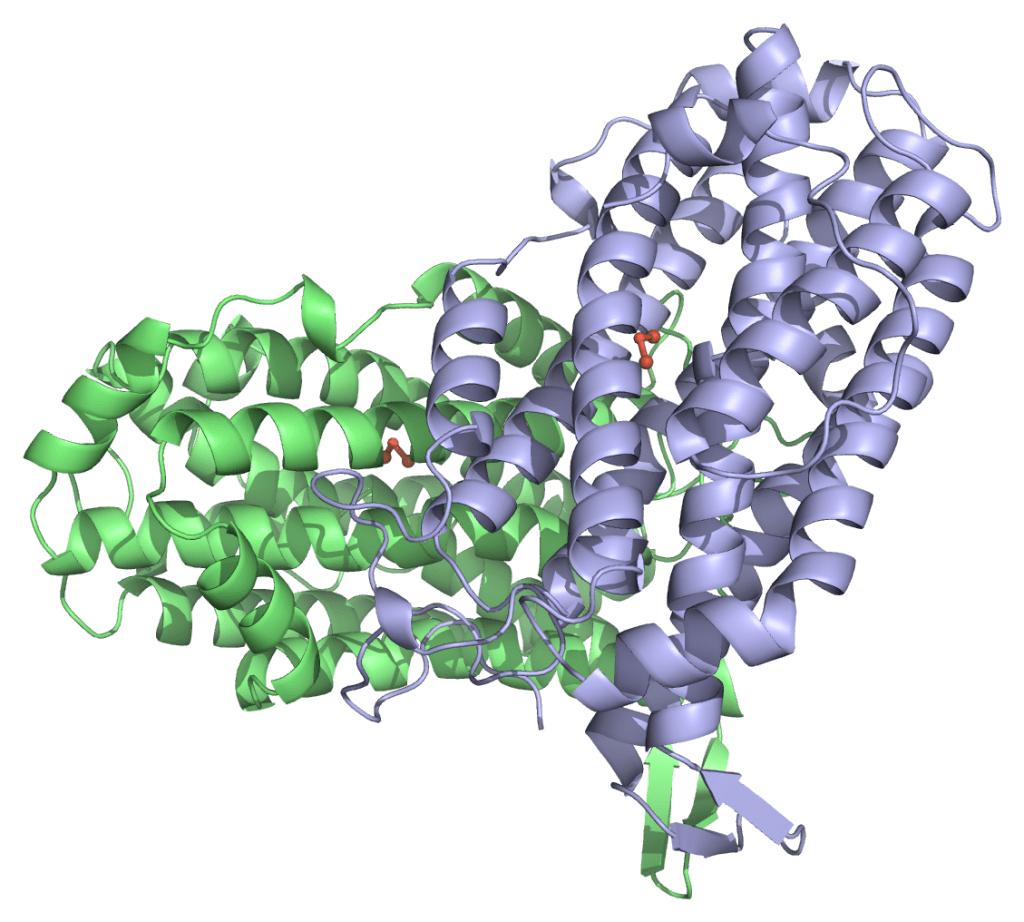
Image 2. Superposition de l’état radical de MfR2 avec des structures représentatives de R2a et R2b. Les structures ont été alignées à l’aide de PyMOL et sélectionnées pour contenir un centre di-métallique et avoir soit de l’eau dans un rayon de 2 Å autour de w1, soit le groupe ε-ammonium de K213, ou les deux (ce qui est le cas uniquement pour 1kgn). Les structures R2a sont montrées en orange, R2b en violet, et la structure radicale de MfR2e est représentée en cyan. D88, Y126 et K213 de la structure radicale de MfR2e sont présentés en bâtonnets, et les résidus homologues dans toutes les autres structures sont représentés en lignes. Les métaux sont représentés par de grandes sphères, et les molécules d’eau par de petites sphères. Les ID PDB des structures sélectionnées sont les suivantes : R2a : 1w68, 1mxr, 7bet ; R2b : 3mjo, 1kgp, 1kgn, 1uzr, 3n37, 2r2f, 6qob, 6tqy, 6qo7, 4bmo, 4bmu, 4bmt.
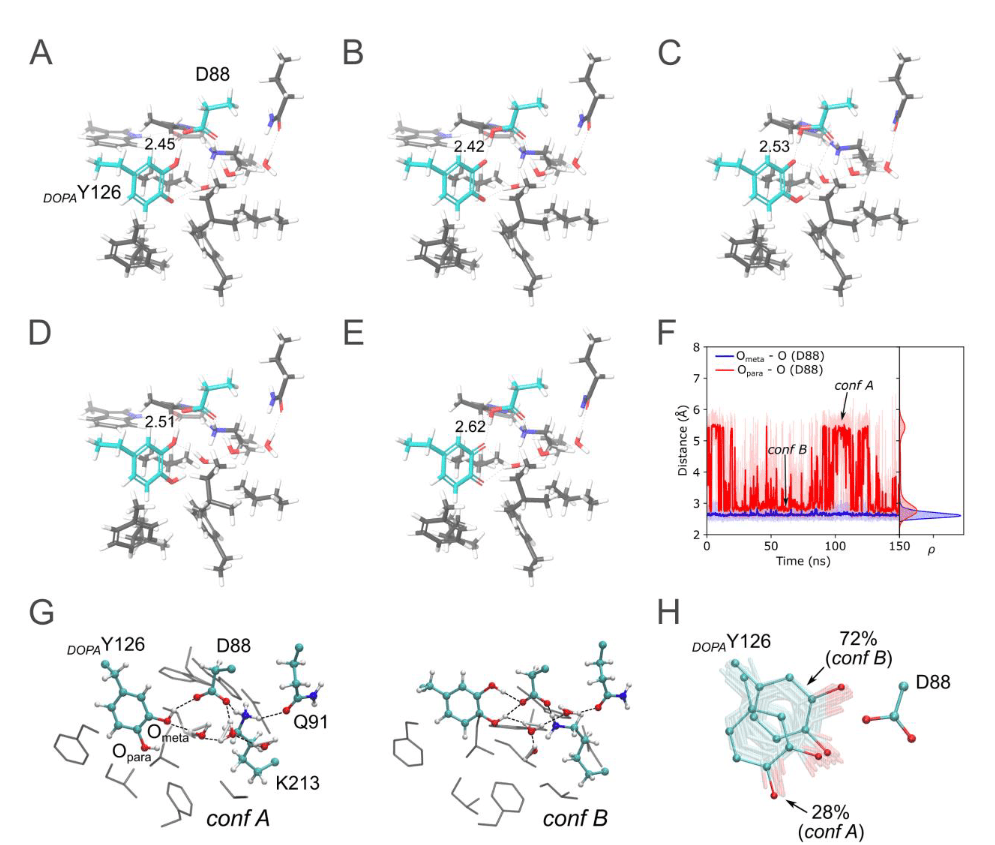
Image 3. Rationalisation théorique des états radical et radical-lost. La courte liaison hydrogène entre DOPAY126 et D88 (résidus colorés en cyan) suggère un état radical (A-E). Sur la base de calculs quantiques sur la structure XFEL, la liaison hydrogène expérimentalement observée entre DOPAY126 et D88 peut être reproduite par un état radical de DOPA avec le radical situé sur le para-O (A) et (B). Un transfert de proton entre DOPAY126 et D88 résulte en un état radical DOPA neutre (A) et chargé négativement (B), avec une énergie similaire. Un état radical DOPA avec le caractère radical sur le méta-O, et D88 protoné, montre une liaison hydrogène plus longue (c). Une DOPA neutre forme une liaison hydrogène plus longue avec D88 (D). L’état quinone oxydé à deux électrons forme une liaison hydrogène plus longue avec D88 que celle observée expérimentalement (E). Les simulations de dynamique moléculaire (MD) à partir de la structure XFEL en état radical suggèrent que DOPAY126, ayant perdu le radical, se déplace dans la position trouvée dans la structure cristalline radical-lost (F à G). L’état conformationnel dominant de DOPAY126 (conf B) forme deux liaisons hydrogène avec D88, tandis que conf A ne forme qu’une liaison hydrogène avec D88. Évolution temporelle des atomes d’oxygène para et méta de DOPAY126 avec D88 pendant la MD (F). Instantanés des deux conformations de DOPAY126 et de leur interaction avec les résidus protéiques environnants et les molécules d’eau (G). Échantillonnage conformationnel de DOPAY126 pendant la MD (H). Les atomes d’azote et d’oxygène sont indiqués en bleu et rouge, respectivement. Les carbones sont représentés en gris ou cyan. Les interactions par liaison hydrogène sont indiquées par des lignes pointillées noires, et les distances O–O sont en Å.
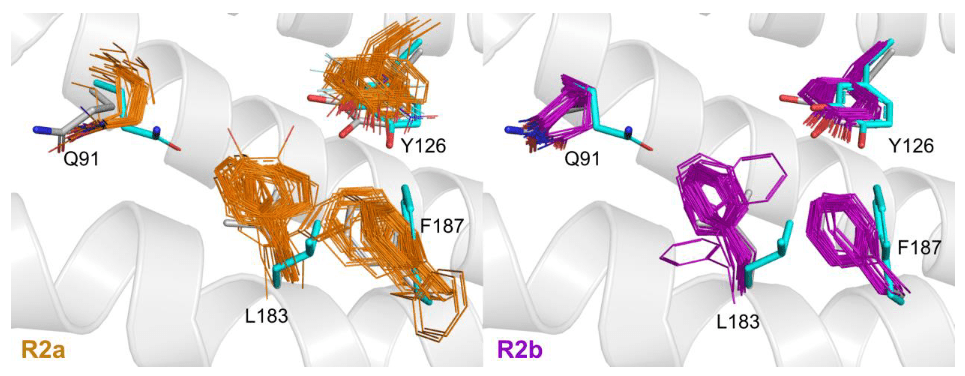
Image 4. Comparaison des structures MfR2e avec toutes les structures publiées de R2a et R2b dans le PDB avec une résolution de 2,5 Å ou meilleure. Toutes les structures ont été alignées avec PyMOL sur la structure à l’état fondamental, et les résidus équivalents à Q91, Y126, L183 et F187 de MfR2e sont représentés en lignes ; les structures R2a sont indiquées en orange du côté gauche et les R2b en violet du côté droit. Les structures radical et à l’état fondamental de MfR2e sont représentées en bâtonnets cyan et gris, respectivement. Pour plus de clarté, seules les hélices de MfR2e à l’état fondamental sont représentées en arrière-plan en tant que structure en ruban. Les résidus Q91, L183 et F187 présentent des conformation uniques dans la structure radicale de MfR2e par rapport à toutes les autres structures. Les PDB IDs des structures alignées sont pour R2a : 6sf5, 3vpn, 3vpo, 3olj, 1h0n, 1h0o, 1w68, 1w69, 1xsm, 4djn, 1piy, 1piz, 1pj0, 1pj1, 1pm2, 1jpr, 1jqc, 1mxr, 1pfr, 1pim, 1piu, 1r65, 1rib, 1rnr, 1rsr, 1rsv, 2av8, 1yfd, 1biq, 5ci0, 5ci1, 5ci2, 5ci3, 5ci4, 1smq, 2o1z, 1xik, 2xof, 7bet, 7ai8, 7ai9, 6zjk, 7aik, 7ail, 7q39,7q3c ; et pour R2b : 6tqv, 6tqw, 6tqx, 6tqy, 6tqz, 6qo5, 6qo7, 6qo8, 6qo9, 6qob, 4bmq, 4bmr, 4bmt, 4bmu, 3mjo, 3n37, 3n38, 3dhz, 1kgn, 1kgo, 1kgp, 1oqu, 1r2f, 1uzr, 2r2f, 4dr0, 4n83, 4m1f, 2bq1, 6mw3, 4bmo, 4bmp, 3n39, 3n3a, 3n3b, 7z3d, 7z3e.

Amelia Maheu
Annonces PARTENAIRES